
 العربية
العربية Español
Español 中文
中文 Deutsch
Deutsch Français
Français Português
Português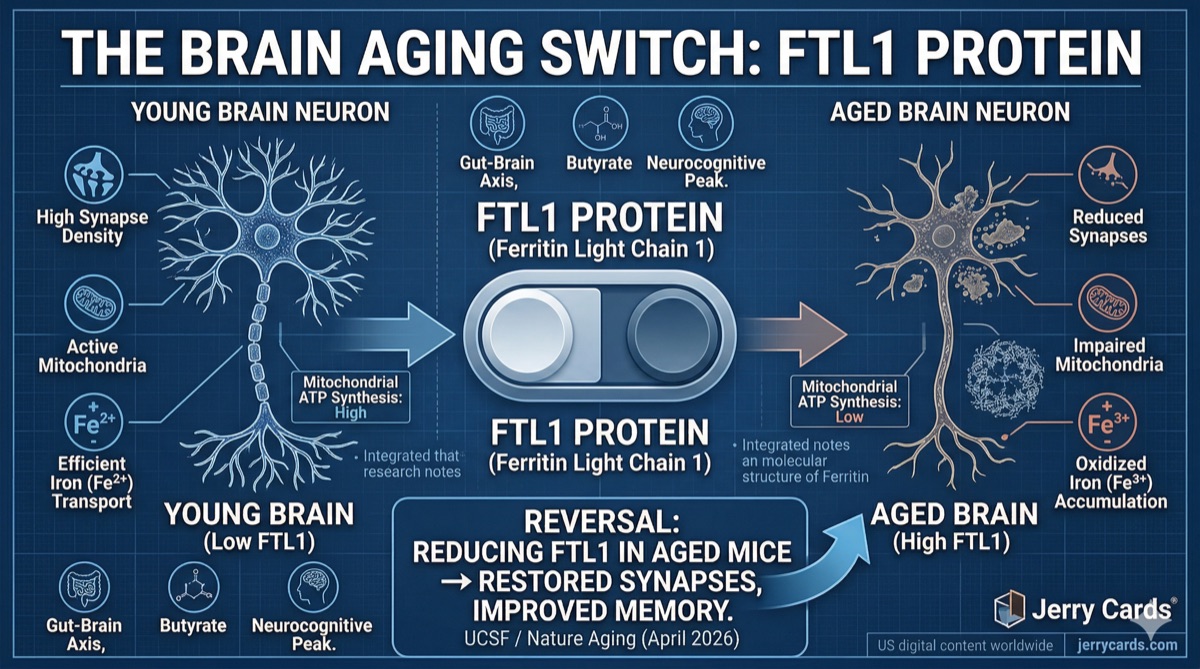
What if brain aging isn’t inevitable — but is driven by a single protein that can be dialed up or down like a switch?
A team at the University of California, San Francisco (UCSF), led by Saul Villeda, PhD (Associate Director of the Bakar Aging Research Institute), has identified exactly that: a protein called FTL1 (ferritin light chain 1) that accumulates in aging brains and directly drives cognitive decline. Published in Nature Aging.
The most remarkable finding: reducing FTL1 in old mice reversed the damage. Synapses regrew. Memory improved. As Villeda put it: "It is truly a reversal of impairments. It’s much more than merely delaying or preventing symptoms."
1. The Discovery: One Protein, Consistently Elevated
The UCSF team used two independent methods to compare the hippocampus of young (3-month-old) versus aged (18-month-old) mice:
| Method | What It Measures | Finding |
| RNA Sequencing | Gene expression levels in hippocampal neurons | FTL1 mRNA significantly elevated in aged mice |
| Mass Spectrometry | Protein levels at synapses | FTL1 protein significantly elevated at aged synapses |
FTL1 was the only protein consistently increased with age across both datasets. Out of thousands of genes and proteins screened, this one kept appearing. That’s a powerful statistical signal.
What is FTL1? Ferritin light chain 1 is a component of the ferritin complex — the protein shell that stores iron inside cells. Iron is essential for brain function (it’s needed for neurotransmitter synthesis and mitochondrial energy production), but it must be tightly regulated. Too much free iron, especially in its oxidized form (Fe³⁺), is toxic. FTL1 is part of the system that manages this balance — but in aging, it appears to malfunction.
2. The Bidirectional Proof: Young → Old, Old → Young
What makes this study exceptionally strong is the bidirectional design — they tested both directions of the switch.
2.1 Making Young Brains Old
Researchers used neuron-specific viral vectors to artificially increase FTL1 in the hippocampus of young adult mice. The results were striking:
- Fewer dendritic branches — neurons became structurally simpler, resembling aged neurons
- Reduced synaptic density — fewer connections between brain cells
- Impaired long-term potentiation (LTP) — the cellular mechanism of learning and memory was weakened
- Cognitive deficits — young mice with elevated FTL1 performed like old mice on memory tests
Brain cells engineered to produce high FTL1 developed "simplified structures, forming short, single extensions" — the hallmark of aged neurons.
Translation: Increasing one protein was sufficient to make a young, healthy brain look and function like an old one. The "aging switch" is real, and FTL1 is the toggle.
2.2 Making Old Brains Young
The reverse experiment is where the study becomes extraordinary. Researchers used three independent methods to reduce FTL1 in the hippocampus of aged mice:
| Method | How It Works |
| RNA Interference (RNAi) | Small RNA molecules silence FTL1 gene expression |
| CRISPR-Cas9 | Gene editing to knock out FTL1 in targeted neurons |
| Conditional Knockout | Genetic engineering to remove FTL1 specifically in adult neurons |
All three approaches produced the same result:
- Enhanced dendritic complexity — neurons grew more branches, more connections
- Restored synaptic markers — NR2A (NMDA receptor subunit), AMPA receptors, and synapsin levels returned to youthful levels
- Improved memory performance — old mice performed significantly better on cognitive tests
The Quote: "It is truly a reversal of impairments. It’s much more than merely delaying or preventing symptoms." — Saul Villeda, PhD, UCSF
3. The Mechanism: Iron, Mitochondria, and Energy Collapse
The complete signaling cascade from FTL1 accumulation to cognitive decline:
Step 1: FTL1 protein accumulates in hippocampal neurons with age
Step 2: Excess FTL1 alters labile iron oxidation states → Fe²⁺ (usable) converts to Fe³⁺ (oxidized, toxic)
Step 3: Oxidized iron (Fe³⁺) interferes with mitochondrial electron transport chain
Step 4: ATP synthesis drops — neurons can’t produce enough energy
Step 5: Energy-starved neurons can’t maintain synaptic connections
Step 6: Dendritic branches retract, synapses wither, LTP impaired
Step 7: Memory and learning decline
3.1 The Iron Paradox
Iron is essential for brain function. Every neurotransmitter synthesis pathway, every mitochondrial energy cycle, requires iron. But iron is also a redox-active metal — it easily switches between Fe²⁺ (ferrous, useful) and Fe³⁺ (ferric, harmful).
In young brains, the ferritin complex (including FTL1) keeps iron safely stored and in the right oxidation state. In aging brains, FTL1 overaccumulates, and the balance tips toward Fe³⁺ — which generates reactive oxygen species (ROS) that damage mitochondrial membranes and DNA.
This connects to a broader theme in aging research: iron dysregulation is increasingly recognized as a driver of neurodegeneration, not just in aging but in Alzheimer’s, Parkinson’s, and other diseases.
4. The Metabolic Rescue: NADH Supplementation
Perhaps the most clinically exciting finding: the aging effects of FTL1 can be prevented without reducing FTL1 itself.
Using neuronal nuclei RNA sequencing, the team detected that FTL1 accumulation specifically disrupted ATP synthesis pathways. When they supplemented neurons with NADH (nicotinamide adenine dinucleotide, a key cofactor in mitochondrial energy production), the pro-aging effects of FTL1 were blocked — even with FTL1 still elevated.
The Implication: You may not need to target FTL1 directly (which requires gene therapy or ASO treatment). Boosting mitochondrial metabolism — supporting the energy supply chain — may be sufficient to counteract FTL1’s damage. This opens the door to metabolic interventions (NAD+ precursors, exercise, dietary strategies) as practical neuroprotective approaches.
5. The Connection to Our Resistant Starch Series
Regular readers will notice a direct parallel with our Resistant Starch series:
| Intervention | Mechanism | Brain Effect |
| Reduce FTL1 | Restore mitochondrial ATP production in hippocampal neurons | ↑ Synapses, ↑ Memory, ↑ LTP |
| NADH supplementation | Directly boost mitochondrial energy production | Prevent FTL1’s pro-aging effects |
| Resistant Starch → Butyrate | Butyrate enters neurons, boosts mitochondrial function, increases BDNF | ↑ Synapses, ↑ Memory, ↓ Neuroinflammation |
Different entry points, same target: keeping hippocampal neurons energized enough to maintain their synaptic connections. FTL1 research attacks the problem from the iron/mitochondria angle; resistant starch attacks it from the gut-brain/butyrate angle. Both converge on neuronal energy as the key to cognitive longevity.
6. Limitations and Honest Caveats
- Mouse model: All experiments were in mice. No human trials of FTL1 reduction have been conducted.
- Iron is essential: Reducing FTL1 too aggressively could disrupt normal iron storage, potentially causing other problems. The dose-response relationship is not yet characterized.
- ASO delivery: Getting antisense oligonucleotides or CRISPR into the human brain at therapeutic doses remains a major bioengineering challenge.
- NADH supplementation: The specific compound and dose used in the study were for cell culture, not oral supplementation. NAD+ precursors (NMN, NR) exist as supplements but their ability to cross the blood-brain barrier at relevant doses is still debated.
- Correlation vs causation in humans: While FTL1 is elevated in aging mouse brains, the human data is less complete. It’s plausible but not yet proven that the same switch operates identically in humans.
7. The Bottom Line
For the first time, researchers have identified a single protein that:
- Accumulates consistently in the aging hippocampus (the only one to do so across both RNA and protein datasets)
- Directly causes cognitive decline when elevated in young brains (bidirectional proof)
- Can be reversed in old brains using three independent methods
- Operates through a known mechanism (iron oxidation → mitochondrial dysfunction → synapse loss)
- Can be metabolically bypassed (NADH supplementation prevents the damage even with FTL1 present)
The brain aging switch is real. And for the first time, we know how to flip it back.
Sources:
- Remesal et al., Nature Aging: Targeting iron-associated protein Ftl1 in the brain of old mice improves age-related cognitive impairment
- UCSF: This Protein Slows the Aging Brain, and We Know How to Counter It
- ScienceDaily: Scientists found a protein that drives brain aging — and how to stop it
- ScienceAlert: Switching Off One Crucial Protein Appears to Reverse Brain Aging